Técnicas de estudio del semen
Esterilidad de origen genético o cromosómico.
Algunas anomalías genéticas y cromosómicas pueden ser causa de esterilidad o infertilidad. En estos casos es necesario realizar estudios específicos.
¿Por qué las anomalías genéticas o cromosómicas pueden ser causa de esterilidad?
- Porque alteran la producción de espermatozoides
- Porque alteran el transporte de los espermatozoides
- Porque pueden dar lugar a la producción de espermatozoides con un contenido cromosómico anómalo
¿Cuáles son las anomalías genéticas o cromosómicas relacionadas con la esterilidad?
Si bien existen muchas otras, las que con mayor frecuencia se relacionan con la esterilidad son:
- Anomalías cromosómicas constitucionales
- Anomalías limitadas a las células germinales
- Microdelecciones del cromosoma Y
- Mutaciones en el gen CFTR de la fibrosis quística (Cistyc Fibrosis Transmembrane Regulator)
¿Qué son las anomalías cromosómicas constitucionales?
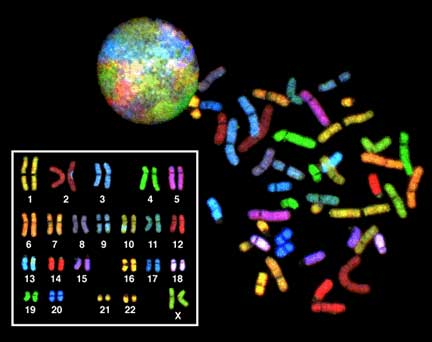
Son las que afectan a los cromosomas de las células somáticas humanas, aunque pueden ser muy complejas, básicamente distinguimos dos tipos: numéricas (pérdida y/o ganancia de uno o varios cromosomas completos que pueden afectar tanto a los autosomas como a los cromosomas sexuales) y estructurales (implican cambios en la estructura de uno o varios cromosomas).Ambos tipos pueden darse simultáneamente. Las células somáticas humanas tienen 46 cromosomas: 22 pares de cromosomas homólogos o autosomas (los cromosomas 1 a 22) y dos cromosomas sexuales. A esto se le llama el número diploide . Las mujeres tienen dos cromosomas X (46, XX) mientras que los varones tienen un X y un Y (46, XY). Generalmente, la pérdida de cromosomas tiene mayor repercusión en un individuo que la ganancia, aunque ésta también puede tener consecuencias serias. Y la pérdida o ganancia de un autosoma tiene consecuencias más graves que la de un cromosoma sexual.
¿Son frecuentes este tipo de anomalías en pacientes estériles?
La incidencia de anomalías cromosómicas constitucionales es unas 13 veces superior en la población de pacientes estériles que en la población general. Siendo más frecuente la alteración de los cromosomas sexuales que la de los autosomas. Por otro lado en los pacientes estériles o infértiles, la incidencia de anomalías cromosómicas constitucionales es inversamente proporcional al recuento espermático, por tanto cuanto más bajo es el número de espermatozoides en el seminograma, más riesgo de padecer una de estas anomalías. Su incidencia en pacientes estériles oscila entre el 2% en normozoospermias al 13% en azoospermias.
¿Cómo puedo saber si padezco una alteración cromosómica?
Mediante la realización de un estudio de cariotipo en una muestra de sangre.
¿En qué casos está indicado el estudio de cariotipo?
El estudio de cariotipo debería realizarse en todo paciente que consulte por esterilidad, siendo imprescindible en los casos de azoospermia, oligozoospermia severa (con o sin asteno y/o teratozoospermia), normozoospermia con abortos recurrentes y en especial en pacientes subsidiarios de tratamiento con fecundación “in vitro” con microinyección intracitoplásmica (FIV-ICSI).
¿Si el cariotipo es normal, ya no es necesario hacer más estudios cromosómicos?
Dependiendo de las características clínicas y analíticas, algunos pacientes deberán someterse a otros estudios. Los pacientes con cariotipo normal pueden presentar anomalías de la meiosis, limitadas a la línea germinal (anomalías en el apareamiento de los cromosomas homólogos, desinapsis, asinapsis). Estas anomalías darán lugar a espermatozoides aneuploides, que en caso de fecundación del oocito serán responsables de la formación de embriones inviables.
¿Cómo se puede saber si mis espermatozoides tienen anomalías en la carga genética?
 En los últimos años, el análisis de los cromosomas se ha beneficiado de la aparición de una técnica que combina la citogenética y el estudio molecular, se trata de la hibridación in situ fluorescente (FISH). En una muestra de semen puede realizarse la FISH de espermatozoides. Sin embargo, las técnicas actuales sólo permiten analizar 5 de los 23 cromosomas (cromosomas 13, 18, 21, X, Y). Por tanto, aunque puede ser una técnica útil si existen anomalías en estos cromosomas, el estudio resulta incompleto. Por esta razón, preferimos realizar un estudio de meiosis, ya que el mismo proporciona información de todos los cromosomas.
En los últimos años, el análisis de los cromosomas se ha beneficiado de la aparición de una técnica que combina la citogenética y el estudio molecular, se trata de la hibridación in situ fluorescente (FISH). En una muestra de semen puede realizarse la FISH de espermatozoides. Sin embargo, las técnicas actuales sólo permiten analizar 5 de los 23 cromosomas (cromosomas 13, 18, 21, X, Y). Por tanto, aunque puede ser una técnica útil si existen anomalías en estos cromosomas, el estudio resulta incompleto. Por esta razón, preferimos realizar un estudio de meiosis, ya que el mismo proporciona información de todos los cromosomas.
¿Qué es la meiosis?
La meiosis es el proceso por el cual las células madre precursoras de los espermatozoides dividen su carga genética a la mitad, y pasan de tener 46 cromosomas a tener 23. Durante este proceso se pueden producir alteraciones que darán lugar a espermatozoides con una dotación cromosómica anómala y podrán provocar esterilidad, abortos o fracasos en las técnicas de FIV.
¿Cuándo está indicado el estudio de la meiosis?
La incidencia de anomalías en la meiosis en pacientes estériles es del 4 al 8%. Sin embargo, en pacientes con recuentos espermáticos muy bajos, (oligozoospermia inferior a 1.5 millones de espermatozoides móviles /ml) se incrementa hasta el 18 %.
Así mismo, en 60 pacientes con semen normal pero que presentaban abortos recurrentes o sucesivos fracasos en FIV (mala calidad embrionaria, baja tasa de fecundación, no gestación), la incidencia de anomalías en la meiosis fue del 45%.
¿Cómo se estudian las alteraciones de la meiosis?
Desde principios de los años 80, los estudios de meiosis se incluyen en los protocolos de estudio de la infertilidad masculina. Se llevan a cabo a partir del análisis de tejido del testículo obtenido mediante una biopsia testicular. Actualmente también se puede estudiar en espermatozoides de eyaculado.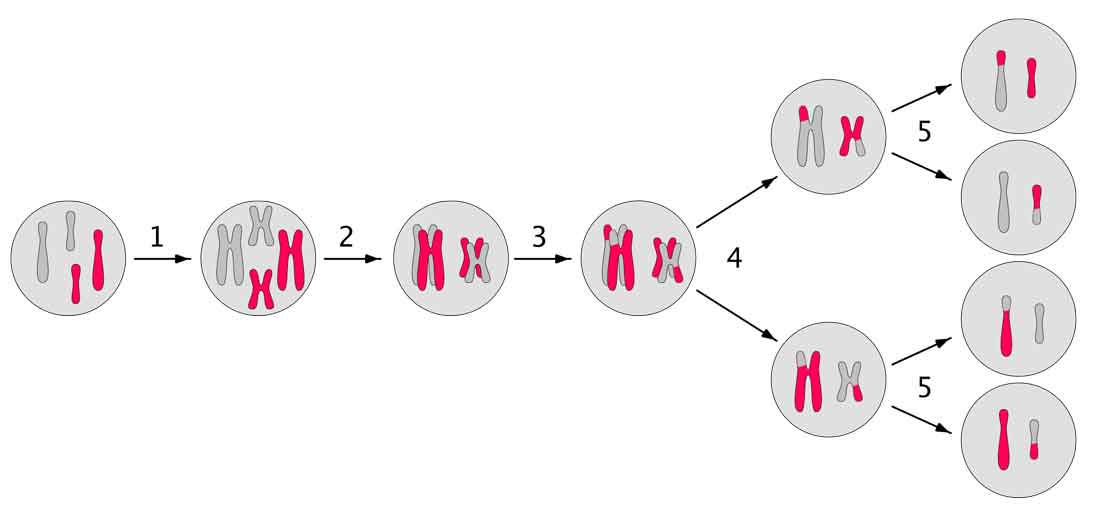
¿En que consiste la biopsia testicular?
Es una pequeña intervención quirúrgica que se realiza bajo anestesia local (sólo del testículo) y de forma ambulatoria (no requiere ingreso). A través de una pequeña incisión de aproximadamente un centímetro en la raíz de las bolsas del testículo se inciden las diferentes capas que rodean al testículo hasta llegar al mismo, para extraer una pequeña muestra. Las diferentes capas del testículo, así como la piel, son suturadas con puntos reabsorbibles (se caen solos sin precisar ser extraídos). Finalizada la intervención el paciente puede regresar a su domicilio. En ocasiones pueden presentarse molestias durante el postoperatorio, que ceden habitualmente con la administración local de hielo y con analgésicos.
¿Tienen tratamiento las alteraciones del cariotipo o de la meiosis?
Estas alteraciones no tienen tratamiento y suponen un mal pronóstico de la fertilidad de quien las padece. No quiere decir que los individuos que las padecen no puedan llegar a producir un mínimo porcentaje de espermatozoides normales cromosómicamente, pero no existe ninguna técnica que nos permita separar los espermatozoides equilibrados cromosómicamente de los que no lo están. Por lo que normalmente se obtendrán un elevado porcentaje de embriones inviables. Puede mejorarse el pronóstico de fertilidad seleccionando los embriones mediante técnicas de Diagnóstico Genético Preimplantatorio (DGP).
¿Qué son las microdelecciones del cromosoma Y?
 El cromosoma Y es el principal determinante de la diferenciación sexual masculina. El cromosoma Y contiene genes importantes para la producción de espermatozoides (espermatogénesis), desde 1976 se conoce que un 0.5% de los varones infértiles presentan deleciones (pérdida del material genético de un cromosoma) del cromosoma Yq (brazo largo del cromosoma Y)). En base a estos resultados se propuso la existencia de un factor de azoospermia en Yq (AZF; Azoospermia Factor). A través de técnicas de análisis molecular basadas en PCR (Polymerase Chain Reaction) fue posible subdividir al cromosoma Y en las regiones AZFa, AZFb y AZFc atendiendo a la región del cromosoma Y que se microdeleciona en estos pacientes. Posteriormente ha sido posible aislar una familia de genes (DAZ; Deleted in Azoospermia) localizados en la región AZFc.
El cromosoma Y es el principal determinante de la diferenciación sexual masculina. El cromosoma Y contiene genes importantes para la producción de espermatozoides (espermatogénesis), desde 1976 se conoce que un 0.5% de los varones infértiles presentan deleciones (pérdida del material genético de un cromosoma) del cromosoma Yq (brazo largo del cromosoma Y)). En base a estos resultados se propuso la existencia de un factor de azoospermia en Yq (AZF; Azoospermia Factor). A través de técnicas de análisis molecular basadas en PCR (Polymerase Chain Reaction) fue posible subdividir al cromosoma Y en las regiones AZFa, AZFb y AZFc atendiendo a la región del cromosoma Y que se microdeleciona en estos pacientes. Posteriormente ha sido posible aislar una familia de genes (DAZ; Deleted in Azoospermia) localizados en la región AZFc.
¿En que casos está indicado el estudio de las microdelecciones del cromosoma Y?
Las microdeleciones del brazo largo del cromosoma Y afectan aproximadamente a un 10% de los pacientes azoospérmicos y a un 15% de los pacientes con oligozoospermia idiopática severa. La mayoría de microdeleciones se producen en la región AZFc y afectan al gen DAZ, siendo éste el principal candidato a explicar la azoospermia de estos pacientes. El estudio se realiza mediante un análisis de sangre.
¿Tienen tratamiento las microdeleciones del cromosoma Y?
No. Los pacientes oligozoospérmicos severos con microdeleción del cromosoma Y son estériles, pero actualmente a través de la técnica de ICSI es posible que tengan descendencia. Incluso en muchos de los pacientes azoospérmicos con microdeleción de Yq es posible recuperar quirúrgicamente espermatozoides del testículo mediante TESA (Testicular Sperm Aspiration) o TESE (Testicular Sperm Extraction) con los que proceder a ICSI. La deleción completa y simultánea de las tres regiones (AZFa, AZFb y AZFc) parece comportar un valor pronóstico negativo ya que en todos los casos se asocia a una ausencia total de espermatozoides. Todos pacientes con una microdeleción tratados con éxito a través de ICSI transmiten la microdeleción y la esterilidad a su descendencia masculina, pero no a su descendencia femenina.
¿Qué son las mutaciones del gen CFTR de la fibrosis quística?
La fibrosis quística (FQ) es la enfermedad autosómica recesiva más frecuente en la raza caucásica, con una prevalencia de uno en 2000-2500 recién nacidos. Una persona de cada 25 son portadoras de esta mutación. Más del 95% de hombres con FQ son estériles. El gen cuya mutación produce la FQ se denomina gen de la FQ o gen CFTR (Cistyc Fibrosis Transmembrane Regulator) según el acrónimo inglés. Se localiza en los brazos largos (q 31) del cromosoma 7. La primera mutación del gen CFTR fue descrita en 1987, habiéndose descrito en la actualidad más de 1517 mutaciones.
¿En qué casos está indicado el estudio de las mutaciones del gen CFTR?
El estudio de las mutaciones del gen CFTR estará especialmente indicado en las azoospermias por ABCD (agenesia bilateral congénita de los conductos deferentes), en la AUCD (agenesia unilateral del conducto deferente) con o sin azoospermia. En la práctica clínica se estudian las mutaciones más frecuentes, unas 33 mutaciones, que representan aproximadamente el 75% de las mutaciones en el gen CFTR en la población española. Además, es preciso en estos casos estudiar el gen CFTR también en la pareja, pues puede ser portadora, de una mutación del gen. De ser así, el embrión podría heredar una mutación del padre; y otra mutación de la madre y presentar Fibrosis Quística. Si la pareja es portadora de una mutación del gen CFTR, está indicado hacer Diagnóstico Genético Preimplantacional (DGP) es decir, estudio del gen de la CFTR en una célula, blastómero, del embrión para conocer si el embrión es afectado o sólo portador de la Fibrosis Quística.
Consulta con el Instituto Europeo de Fertilidad ante los primeros síntomas de infertilidad.
Nuestra clínica y todo el equipo que lo formamos estamos comprometidos con la excelencia y el trato personalizado a cada una de nuestras pacientes.
Cada paciente es única y así enfocamos nuestro trabajo hasta conseguir el embarazo con éxito.

Ginecólogo especialista en Reproducción Asistida
Miembro de la Sociedad Española de Fertilidad
